
-
 Shifa Miyaji
Shifa Miyaji
- October 28, 2021
The medical device industry is one of the largest industries in healthcare, and its growth is exponentially on the rise. The market size for global medical devices was valued at US$ 447.63 Billion in 2019, and by 2027, its value is expected to be US$ 671.49 billion.
The latest medical devices are constantly driven by new technologies and innovations, requiring in-depth research, stringent testing and bio-technical mastery. Yet, this entire process is just the tip of the iceberg. When it comes to launching the final product or device in the market (both local and foreign), there is a long list of stringent regulations that companies must follow for their product to be eligible for use. These involve major administrative, linguistic and regulatory requirements where errors or inaccuracy can cost you a lot!
This immediately calls upon a dire need for multilingual translation services. Medical device translation is one of the most complex and demanding types of translation. And you need to be doing it 100% right to save lives.
Firstly, let’s take a deeper look at what qualifies as a medical device. Here, we shall discuss the medical device product cycle, the changes brought by the Medical Device Regulation in the European Union and best practices for accurate translations.
How is a medical device defined?
According to the FDA (US Food & Drug Administration), a medical device is used to diagnose, prevent, or treat a medical disease or condition without causing any chemical reaction on any part of the body.
There are mainly 3 classes of medical devices, but they can slightly vary depending upon the country or the region, as is visible below.
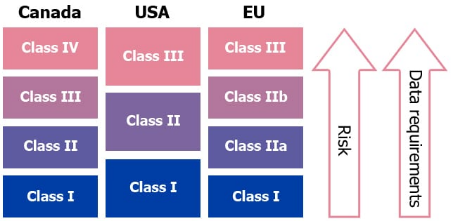
From Product Development To Market Launch
After a detailed market analysis and research about its market demand, a team of biotech experts and innovators finalise the concept and design of the medical device. The next few stages involve multiple rounds of clinical trials, testings, feedbacks and reviews until the device is error-free, safe and effective and its operability is crystal clear.
Once the medical devices are manufactured, companies send forward their products to users and medical institutions on a global scale. Most countries and regions have their own medical device regulations for market entry. These are highly regulated – any content associated with the device is closely scrutinised before entering a new market. As such, these documents and their medical translations must be accurate.
Check out this video that explains the difference in regulations and regulatory bodies in the USA and the European Union.
Some of the materials that regulatory bodies typically require to translate include:
- packaging and labels
- marketing materials
- instructions for use
- software and user interface
In general, you need to translate all documents such as clinical trials, administrative records, and quality management system certificates that regulators will use to assess a device and approve whether it can be imported and distributed to the target market.
These requirements make medical device translation integral in the approval process.
As over half a million different medical devices are available in the European Union alone, the European Union (EU), in 2017, published a comprehensive list of requirements for medical device companies to produce and commercialise their medical devices being distributed to the EU market. An essential element in this is how companies are required to handle their translation submittals.
A Brief Introduction About The EU MDR
The European Medical Device Regulation (MDR) is the latest set of regulations governing the production and distribution of medical devices in Europe. Compliance with the regulation is mandatory for medical device manufacturers, importers, and distributors that wish to target the European marketplace.
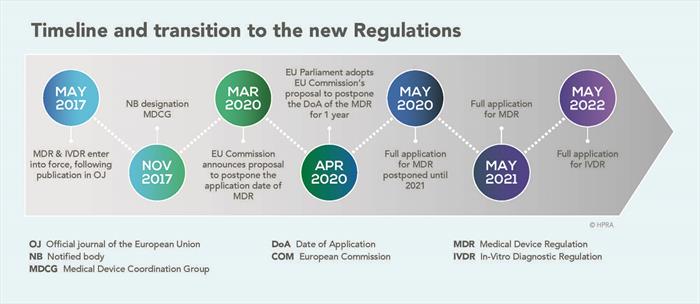
The Medical Device Regulations were established on 5 April 2017. There were many reasons which required the previous MDD (Medical Devices Directive) to be updated.
For instance, software as a medical device (SaMD) wasn’t yet evolved, and hence, there were no direct mentions of it in the directive, but now, they’re a substantial part of the medical device industry.
The altering of demographics and the growing push for transparency of medical device technical information to the general public are also some reasons why the MDR was put in place for a more robust EU legislative framework that ensures better protection of public health and patient safety.
The EU Regulations were first published in May 2017. The MDR initially provided for a three-year transition period; however, due to the global outbreak of Covid-19, the application of the MDR was extended and became fully applicable on 26 May 2021.
Compared to its predecessor, the MDD, the new European MDR is less focused on the pre-approval stage of medical device manufacturing. Instead, it promotes a life-cycle approach to medical device regulation.
You can access the entire EU Medical Device Regulation here.
Language, Translations And The EU MDR
With the new EU MDR in place, translations are now even more critical than they were before. Now, manufacturers will be responsible for acquiring them themselves rather than relying on the EU member states for these. Make sure you adhere to the following points that talk about the language aspect of medical devices as part of the EU regulation:
1. All medical device documents need to be translated into all 24 EU languages
The regulation states that to be compliant, all content must be translated into the 24 official languages of the EU member countries where the devices plan on being distributed. The official EU languages are: Bulgarian, Croatian, Czech, Danish, Dutch, English, Estonian, Finnish, French, German, Greek, Hungarian, Irish, Italian, Latvian, Lithuanian, Maltese, Polish, Portuguese, Romanian, Slovak, Slovene, Spanish & Swedish.
2. Translations, in some cases, are required even prior to the CE marking
Each medical device must be certified with a ‘CE’ (Conformité Européenne) mark to demonstrate compliance before it is sent out to the market. Some devices may even require more than one CE mark depending upon whether they are made up of different parts or accessories. Previously, translations came into the picture after a medical device received the CE marking, but now translations of documents like Instructions for Use (IFUs) must be submitted before the CE marking as part of the technical dossier, which is required by the notified bodies for conformity assessment.
3. All translations must be precise and clear
It is crucial to ensure that the end-user—healthcare staff, patients, or associated users – know how to operate any particular device. In particular, the utmost precision is required for medical device translation, as professionals worldwide will need to know the correct way to use the equipment. As such, appropriate and accurate medical device labelling is essential. Hence, choosing translators with expertise in the medical field isn’t enough. They need to be native speakers of the targeted language too.
4. All translated data must be stored in a European database
The MDR also introduced new databases for clinical investigations, product registration, and marketing surveillance – EUDAMED (European Database for Medical Devices). This will function as part of a system of several databases that allows medical device companies, consumers, regulators, and even Notified Bodies to access the latest data on medical devices for sale in Europe. This will ensure that the data in all applicable languages is accessible to be logged and exchanged.
How to Select The Ideal Translation Agency To Prepare You For The EU MDR?
Experienced translation agencies understand and can help you adhere to the new requirements with timely compliance, accuracy and relevant skills. Don’t make the mistake of approaching translated device documents as an afterthought, especially when trying to enter a foreign market.
You know you can’t risk it as lives are at stake when it comes to medical devices. Just remember these simple rules to get quality medical device documentation translations.

Conclusion
The EU MDR website recommends that you wisely select ‘the company or companies that will be developing, potentially producing and ultimately commercialising’ your product. Hence, you must choose a translation company that is highly knowledgeable and experienced in medical translations and the legal and regulatory compliances of the targetted country.
Translate By Humans’ database of global translators with in-depth industry knowledge of medicine and technology, associated legalities, and specific jargon will allow your device to reach international markets and the patients in need.

